
致敬天河应用创新发展—“天河一号”助力新型电池材料的研发
导读:
二次电池作为能源化学重要研究组成部分,在3C电子、电动汽车和储能等方面具有很大的应用前景。
理论计算化学的快速发展,促进了对电极材料反应机理的认识。通过计算机的有效模拟,我们可以发现电极材料固有的性质特征,发现一般规律,进而帮助指导和设计高效的电极材料。
目前,通过理论计算,我们可获悉脱嵌离子在电极材料上的反应位点,了解金属阳离子与电极反应的历程。通过电子结构信息计算,理解离子间的反应方式,明确反应机理。具体模拟手段有分子反应动力学计算表界面作用关系,电解液成分的相互作用,密度泛函理论计算离子脱嵌反应结构,结构性质(表征包括电子态密度、分子表面静电势,轨道能级分布)等。
近两年来,天津超算中心用户南开大学陈军院士课题组基于“天河一号”采用第一性原理计算研究新型电池材料的反应机制,取得一系列研究成果。
成果信息:
发展了分子表面静电势方法预测有机电极材料(有机小分子,聚合物以及有机晶体)锂化过程,相关结果发表在J.Phys.Chem.Lett.(2018)。此外,该方法所得规律在实验工作中得到验证,其相关成果发表在国际顶尖学术期刊Sci.Adv.(2018)和德国应用化学Angew.Chem.Int.Ed.(2017)。
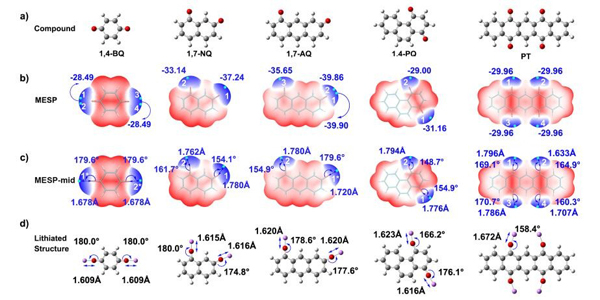
五种有机分子(1,4-BQ,1,4-PQ,1,7-AQ,1,7-NQ和PT)的(a)优化结构,(b)分子表面静电势,(c)分子表面静电势简化图,(d)锂化结构
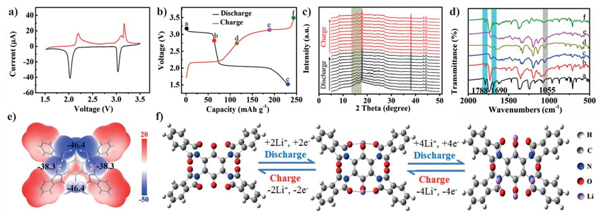
PTB分子嵌锂结构机理图及充放电测试图
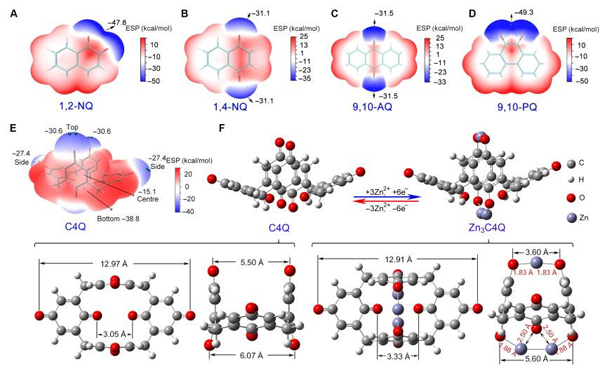
4Q嵌Zn机理图
探究了醌类化合物作为锂离子电池正极材料结构与电化学性质之间的关系。通过对比20种醌化物同分异构体,探究了两种计算方法:考虑电子亲和能和∆A2Li用于设计高电压该类材料,为设计高能量密度的有机正极材料提供理论指导。相关工作发表在Phys.Chem.Chem.Phys.(2018)。
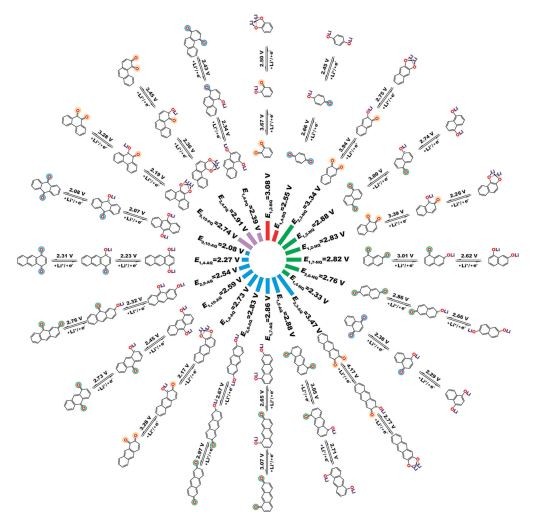
20种不同醌化物的反应机理及平均电压
使用DFT计算模拟对比了硫饱和的二硫化钼纳米带和块体二硫化钼的吸附、扩散以及电子结构性质,发现硫饱和的二硫化钼具有更高的储钠理论容量和钠离子扩散速率。得益于硫饱和的二硫化钼纳米带具有的独特的边缘效应,此发现为设计高性能二硫化钼钠负极材料提供了理论指导,相关工作发表在Chin.J.Chem.(2017)。
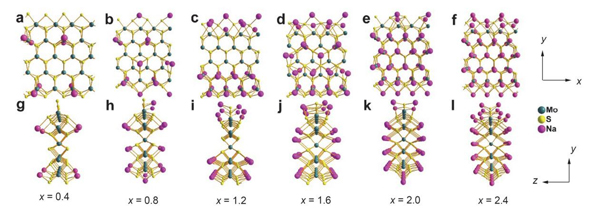
不同的Na在S饱和的MoS2NRs上的吸附构型
计算了石墨烯与6种有机羰基化合物之间吸附作用机制,发现通过官能团修饰改性有机正极材料可形成π-Li-π相互作用。有助于石墨烯与电极材料间的结合,进而起到抑制电极材料溶解和提高电子电导率的作用,有助于合理设计高性能的有机正极材料。相关工作发表在Sci.China.Mater.(2017)。
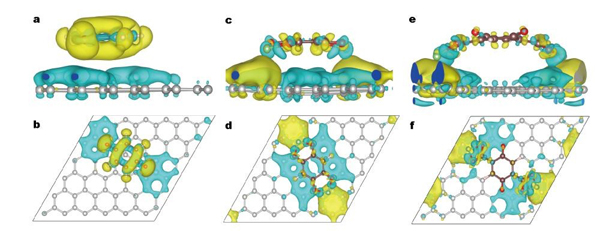
Q/graphene (a,b), BQ-2OLi/graphene (c,d) 以及BQ-2COOLi/graphene (e,f)
差分电荷密度俯视图和侧视图
上述研究工作得到了国家重点研发计划、国家自然科学基金重点项目、教育部创新团队等的支持。论文的计算工作得到了国家超级计算天津中心的大力支持。
论文链接:
https://pubs.acs.org/doi/abs/10.1021/acs.jpclett.8b01123
http://advances.sciencemag.org/content/4/3/eaao1761.short
https://onlinelibrary.wiley.com/doi/full/10.1002/anie.201706604
https://pubs.rsc.org/en/content/articlehtml/2018/cp/c8cp00597d
https://onlinelibrary.wiley.com/doi/abs/10.1002/cjoc.201600744
https://link.springer.com/article/10.1007/s40843-017-9047-0